Vererbung
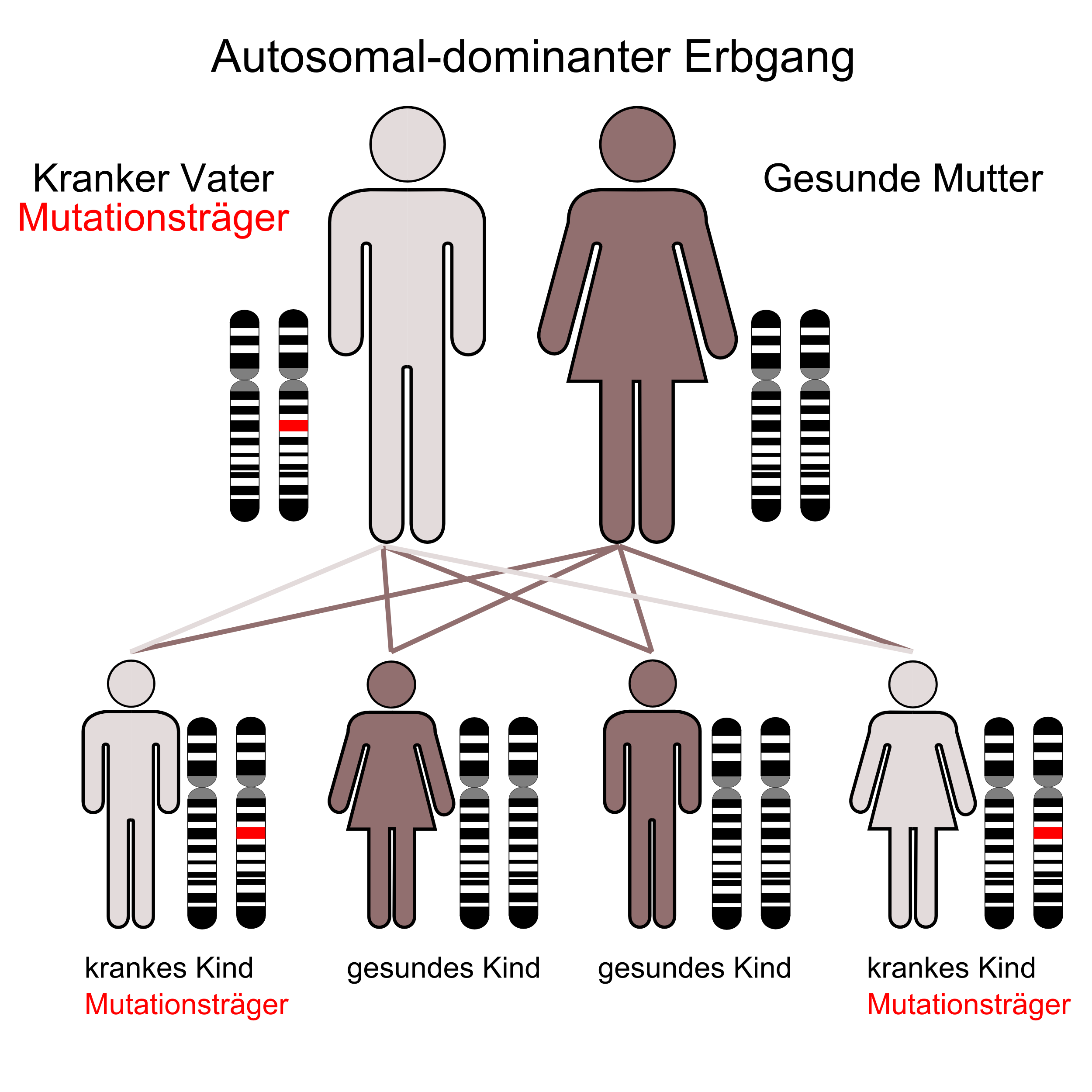
Die Huntington-Krankheit ist eine genetisch bedingte Erkrankung, die autosomal dominant vererbt wird. Autosomal bedeutet: das Gen liegt nicht auf einem Geschlechtschromosom. Deshalb können Männer und Frauen das Gen gleichermaßen erben und damit die Krankheit entwickeln. Dominant bedeutet, dass bereits die Veränderung einer Erbanlage zur Erkrankung führt. In allen Zellen haben wir zwei Erbanlagen (Chromosomen), eine von der Mutter und eine vom Vater. Manche Erkrankungen brechen nur aus, wenn beide Erbanlagen verändert sind. Bei der Huntington-Krankheit genügt es, wenn eine Erbanlage verändert ist. Kinder von Eltern, bei denen ein Elternteil die Gen-Veränderung trägt, haben somit eine 50-prozentige Wahrscheinlichkeit, das Gen zu erben und dann zu erkranken. Das bedeutet aber nicht, dass stets die Hälfte der Kinder in einer Familie oder genau jedes Zweite erkranken wird. Die genannten 50 Prozent sind ein statistischer Wert. Ob ein Kind das Gen erbt oder nicht, bleibt dem Zufall überlassen. Hat ein Kind das Huntington-Gen nicht geerbt, werden weder es noch seine Kinder erkranken, denn die Huntington-Krankheit überspringt keine Generationen. Diese Zusammenhänge zu kennen ist besonders für diejenigen Paare wichtig, die Kinderwünsche haben.
By Kuebi = Armin Kübelbeck [CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0)], via Wikimedia Commons
Gen-Veränderung
Die Gen-Veränderung (Mutation), welche für die Huntington-Krankheit verantwortlich ist, liegt im Gen des Chromosoms 4. Normalerweise werden in diesem Bereich die Nukleinsäuren Cytosin-Adenin-Guanin (CAG) 10 bis 30 Mal wiederholt. Nukleinsäuren sind diejenigen Bausteine, welche die Erbsubstanz bilden. Das veränderte Gen, das die Huntington-Krankheit verursacht, weist jedoch weit mehr CAG-Wiederholungen auf. Sind es mehr als 36, führt dies zur Huntington-Krankheit. Sind es weniger als 30, bricht die Krankheit nicht aus. CAG-Wiederholungen zwischen 30 und 35 stellen einen Graubereich dar, bei dem eine Prognose nicht mit Sicherheit gestellt werden kann. Bei über 60 CAG-Wiederholungen kann die Krankheit bereits vor dem 20. Lebensjahr auftreten. Patienten, die erst nach dem 60. Lebensjahr erkranken, haben meist weniger als 45 Wiederholungen.
Das veränderte Gen wird auch Huntingtin-Gen (nicht Huntington) genannt. Wenn der Vater das kranke Gen vererbt, kann die Anzahl der Wiederholungen bei der Vererbung des Gens auf die Kinder zunehmen. Daraus können ein früherer Krankheitsbeginn und ein schwererer Verlauf resultieren. Wird das Gen von der Mutter vererbt, treten in der Regel keine Änderungen der Wiederholungszahl auf.
In etwa zwei bis fünf Prozent der Fälle finden sich keine Erkrankungen in der Familiengeschichte. Dabei kann es sich um völlig neu entstandene Veränderungen handeln, so genannte Neumutationen. Es kann aber auch sein, dass der Vater eines Huntington-Kranken auf dem Chromosom 30 bis 35 Wiederholungen hat. Die Erkrankung bricht deshalb bei ihm nicht aus. Bei Vererbung auf die Kinder aber hat die Anzahl der Wiederholungen auf über 36 zugenommen und die Kinder entwickeln die Huntington-Krankheit.
Es besteht also eine eindeutige Beziehung zwischen der Anzahl der CAG-Wiederholungen und der Schwere der Erkrankung. Je mehr Wiederholungen vorliegen, desto früher ist mit dem Ausbruch der Erkrankung zu rechnen und desto ungünstiger ist dann die Prognose. Allerdings kann von der Zahl der Wiederholungen nicht direkt auf das Alter geschlossen werden, in dem die Krankheit ausbricht. Selbst bei eineiigen Zwillingen, die identische Erbanlagen tragen, kann die Krankheit zu unterschiedlichen Zeiten und in unterschiedlicher Schwere auftreten. Die stets mit dem Tod endende Krankheit bricht im Durchschnitt zwischen dem 35. und 45. Lebensjahr aus. In seltenen Fällen kann sie auch in der frühen Kindheit oder im höheren Alter auftreten. Bei Kindern kann der Verlauf nur wenige Jahre betragen, bei Menschen im fortgeschrittenen Alter bis zu zwei Jahrzehnte oder mehr. Welche äußeren Faktoren den Beginn und den Verlauf der Krankheit beeinflussen, ist bisher nicht klar.
Auswirkung
Als Folge der Gen-Mutation bildet der Körper einen veränderten Eiweißbaustein. Der nicht veränderte Eiweißbaustein heißt Glutamin. Bei der Huntington-Krankheit tritt dieser Eiweißbaustein häufiger auf und wird daher auch Polyglutamin genannt. Das daraus gebaute Eiweiß ist sehr groß und kann mit sich selbst oder mit anderen Eiweißen verklumpen. Dieses veränderte Eiweiß findet man im Zellkern der Nervenzellen. Die davon betroffenen Nervenzellen verändern sich mit der Zeit, können nicht mehr richtig funktionieren und werden nach und nach ganz zerstört. Dieser Vorgang kann derzeit weder aufgehalten noch umgekehrt werden. Wie das genau vor sich geht, ist noch nicht völlig erforscht. Jedenfalls geschieht dies in einem zentralen Element des Gehirns, dem Striatum. Dies ist zuständig für die Koordination aller Bewegungen, Ordnung aller Informationen, für Emotionen etc.. Bei Schädigung eines solchen Schlüsselelements des Gehirns ist es nachvollziehbar, dass der Ausfall wesentlicher physischer und psychischer Fähigkeiten die Folge sein muss. Durch das Zellsterben nimmt außerdem das Gewicht des Gehirns um bis zu 30 Prozent ab.